2022-09-02今日SH688302股票最新净值和交易情况
海创药业-U(SH688302):
北京8月30日讯昨日晚间,海创药业(688302.SH)披露了2022年半年度报告。今年上半年,海创药业的营业收入为0元,上年同期为0元;归属于上市公司股东的净利润为-1.48亿元,上年同期为-1.48亿元;归属于上市公司股东的扣除非经常性损益的净利润... 网页链接
老柏树也有春天:
老柏与人为善,无意争端。但是侠气还在。真看不惯,装逼的。有钱算个求。
从现在开始,凡是装的,我都要怼。
![[加油]](http://js.xueqiu.com/ugc/images/face/emoji_05_struggle.png)
豪就行了。
挽澜者-垚融:
![[捂脸]](http://js.xueqiu.com/ugc/images/face/emoji_33_face.png)
回复@挽澜者-垚融: 海创药业推31进跌下28就跑出来嘲笑我看50,工厂司机的眼光也就三米!//@挽澜者-垚融:回复@挽澜者-垚融:渔夫山泉11号歪吹芯原股份,今年跌破30明年跌破20,为何你做一辈子工厂司机?难道还不知道原因吗?
每日经济新闻:
每经AI快讯,海创药业(SH 688302,收盘价:35.95元)8月29日晚间发布公告称,经总经理提名、提名委员会审议通过,董事会同意聘任郭宏博士担任公司首席医学官。
海创药业的董事长、总经理均是YUANWEI CHEN(陈元伟),男,59岁,学历背景为博士。
截至发稿,海创药业市值为36亿元。
道达号(daoda1997)“个股趋势”提醒:1. 近30日内无机构对海创药业-U进行调研。更多个股趋势信息,请搜索微信公众号“道达号”,回复“查询”,领取免费查询权限!
每经头条(nbdtoutiao)——面对人口负增长,也许不必如此悲观

(记者 蔡鼎)
免责声明:本文内容与数据仅供参考,不构成投资建议,使用前请核实。据此操作,风险自担。
每日经济新闻
海创药业-U(SH688302):
智通财经APP讯,海创药业(688302.SH)披露2022年半年度报告,该公司研发项目进度持续推进,无产品销售收入,尚未实现盈利,归属于上市公司股东的净亏损1.48亿元,归属于上市公司股东的扣除非经常性损益的净亏损1.64亿元,与上年同期基本... 网页链接
James20162017:
海创药业-U(SH688302)看了很久,重点看了下面两点:
2022年6月经独立数据监查委员会(IDMC)对临床结果审核后判定试验达到方案预设的主要研究终点,并已向国家药品监督管理局药品审评中心递交中国上市前的沟通交流申请;同时,报告期内已完成药物相互作用(DDI),物质平衡(MB)等相关试验。公司正积极开展商业化团队建设、商业化批次生产和质量控制、NDA申报资料撰写等相关工作,争取新药尽快上市并成功实现商业化。如果HC-1119获得批准,将是首款获批上市治疗阿比特龙/化疗后的mCRPC的国产创新药物,有望填补这个治疗领域的空缺市场,解决患者未满足的临床需求。
产品生产准备方面,报告期末,公司位于成都天府国际生物城的募集资金投资项目“研发生产基地建设项目”,已通过四川省外商投资项目备案,已获得环评审查批复、完成节能审查、取得项目用地并签订《国有建设用地使用权出让合同》,取得《建筑工程施工许可证》,研发车间一、研发车间二、生产车间、库房以及生产配套用房等,正在紧张建设中,旨在打造高标准、高质量、高效率的产业化基地,争取早日实现从前期的药物发现、到生产工艺开发与生产规模放大、到GMP体系下的药品制造的全产业链的自主经营,同时,公司遵循上市许可人制度原则(MAH),不断完善符合MAH要求的QA部门和质量系统,确保未来商业化产品符合GMP的要求。
海创药业-U(SH688302):
海创药业2022年半年度董事会经营评述内容如下: 一、 报告期内公司所属行业及主营业务情况说明 (一)主营业务情况 1、主要业务 海创药业是一家基于氘代技术和PROTAC靶向蛋白降解等技术平台,以开发具有重大临床需求的Best-in-class(... 网页链接
海创药业-U(SH688302):
海创药业-U:2022年半年度报告 网页链接
海创药业-U(SH688302):
海创药业-U:2022年半年度报告摘要 网页链接
海创药业-U(SH688302):
海创药业-U:2022年半年度募集资金存放与实际使用情况的专项报告 网页链接
海创药业-U(SH688302):
海创药业-U:中信证券股份有限公司关于海创药业股份有限公司高级管理人员离职暨变更核心技术人员的核查意见 网页链接
海创药业-U(SH688302):
海创药业-U:关于高级管理人员离职暨变更核心技术人员的公告 网页链接
海创药业-U(SH688302):
海创药业-U:第一届监事会第十四次会议决议公告 网页链接
海创药业-U(SH688302):
海创药业-U:独立董事关于公司第一届董事会第二十四次会议相关事项的独立意见 网页链接
海创药业-U(SH688302):
海创药业-U:关于聘任高级管理人员的公告 网页链接
James20162017:
海创药业-U(SH688302)看开拓药业的表现,你能不能争气点
自由之路宝古佬:
1.说一个股票,欧比特,只要跌到8元以下可以无脑买,中长线值得拥有。
2.海创药业短期看好,短线下周逢低可进。
3.下周是化工的天下,化工三傻云天化,兴发集团,湖北宜化,看谁先涨停爆发。
4.长园集团目前价格值得进入,短期10个点能看到。
海创药业-U(SH688302):
同花顺(300033)数据中心显示,海创药业8月22日获融资买入105.21万元,占当日买入金额的11.33%,当前融资余额3309.54万元,占流通市值的3.59%,低于历史40%分位水平。 融资走势表 日期融资变动融资余额8月22日-85.76万3309.54万8月19日-344... 网页链接
空之客:
![[斜眼]](http://js.xueqiu.com/ugc/images/face/emoji_61_xieyan.png)
前几天翻了几篇比较近期关于靶向蛋白降解的reviews,斗胆整理总结一下,大概可以算是review of reviews 引用文献列在最后。
1.技术发展沿革
靶向蛋白降解(TPD)具有治疗性调节蛋白质的潜力,特别是那些已被证明难以用常规小分子靶向的蛋白,它们活性位点很宽、很浅、很难用小分子桥接,还有表面很“光滑”、缺少小分子的结合位点。这些靶点在肿瘤和其他疾病中具有关键作用,且对小分子抑制剂有耐药问题,因而有很大开发潜力。
自文献报道第一个小分子PROTAC以来已有20多年,这项技术已经从学术界走向工业界:2019年首批分子进入临床开发阶段,2020年在雌激素受体(ER)和雄激素受体(AR)两个靶点上完成了POC,TPD领域正在向“难以成药”靶点迈进。

2. 蛋白降解路径
蛋白质稳态(Proteostasis)是指细胞用来维持蛋白质的浓度、构象和亚细胞定位的高度复杂和相互关联的过程,包括很多控制蛋白质的合成、折叠、运输和清除的信号通路。在真核细胞中,受损的蛋白质或细胞器可以被蛋白酶体或溶酶体清除,这两条途径是独立、但又相互联系的。一般来说,蛋白酶体通过泛素-蛋白酶体系统(UPS)来消除短寿蛋白质和可溶性错误折叠蛋白质,溶酶体通过内吞、吞噬或自噬途径来降解长寿蛋白质和不溶性蛋白质聚集体、甚至是整个细胞器和胞内寄生物(例如某些细菌)。


2.1 蛋白酶体途径的靶向蛋白降解
UPS包括蛋白酶体、泛素连接酶和去泛素化酶(DUBs),负责降解受损、未折叠和无用的蛋白质。76个氨基酸残基的泛素蛋白通过赖氨酸异肽键作为翻译后修饰(PTM)连接到蛋白质上,先后涉及三种酶的反应:泛素活化酶(E1)、泛素结合酶(E2)和泛素连接酶(E3),E1消耗ATP与泛素分子结合,然后通过与E2的相互作用将泛素转移到E2,再由E3催化泛素从E2转移到底物上,这三种酶的重复作用导致底物的多重泛素化。根据缀合的泛素分子数目,有8种不同的多泛素链(其中7个赖氨酸残基K6、K11、K27、K29、K33、K48、K63和1个甲硫氨酸残基),其中在哺乳动物细胞中K48和K63键最常见、占比约80%,K48泛素链标记的蛋白通常靶向蛋白酶体进行降解,K63泛素链标记的蛋白在蛋白酶体降解中不起作用、但在调节溶酶体功能和炎症反应中起关键作用。
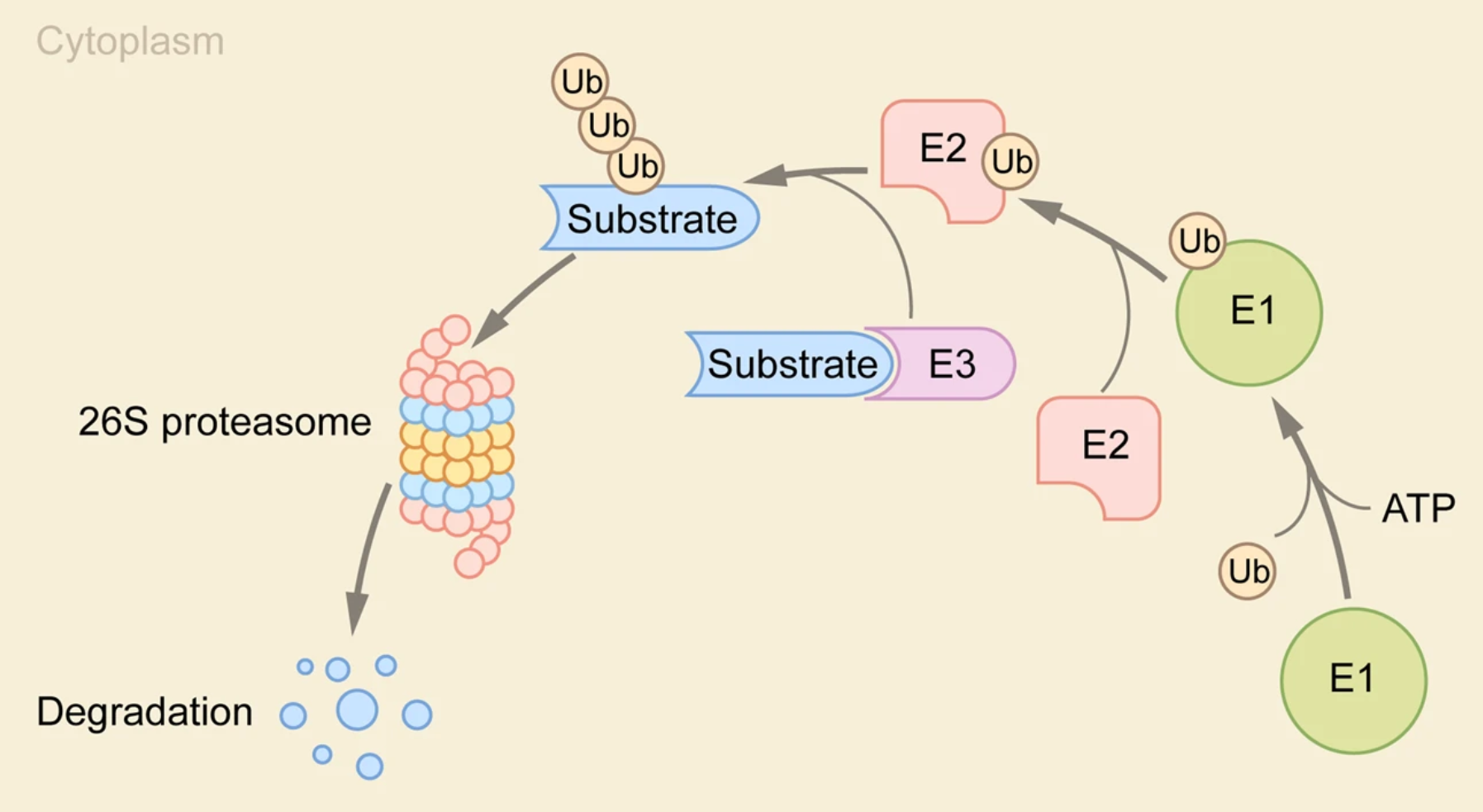
在典型的泛素化通路中,泛素通过E1-E2-E3酶的级联反应与靶蛋白结合。由于E3连接酶负责识别底物,并且其家族的数量大大超过E1和E2,基于UPS的TPD策略利用E3连接酶作为降解的靶向蛋白。通过UPS来降解目标蛋白(POI)主要包括两种技术,即PROTAC和分子胶,以及很多基于PROTAC的技术,如选择性雄/雌激素受体降解剂(SARD/SERD)、疏水性标签(HyT)、TF-PROTAC、双PROTAC等。
2.1.1 PROTAC
PROTAC分子包含E3招募的配体、靶向POI的弹头和连接两个配体的柔性连接子,它能使得POI-PROTAC-E3形成三元复合物,诱导POI的泛素化和随后通过UPS的降解。
与传统的小分子抑制剂相比,PROTAC具有多种优势:1)它极大地扩展了可成药蛋白的范围,目前已经鉴定了4,000多种疾病相关蛋白,其中只有约400种蛋白质成药,许多由于结构复杂、脱靶效应等原因而不能被传统抑制剂靶向;2)传统的小分子抑制剂只阻断蛋白质的部分功能,而PROTAC则会直接降解蛋白质,从而消除其所有功能;3)传统的激酶抑制剂通常在靶点的突变或过表达时会出现耐药性,但PROTAC可以通过降解靶蛋白,将长期选择压力导致的耐药性降至最低;4)PROTAC在亚化学计量和催化方式下具有活性,因而可以在低浓度下发挥作用,从而减少可能的毒副作用。
2.1.2 分子胶
分子胶通过形成三元复合物,来促进两种蛋白质的二聚化或共定位,可以调节多种生物过程,如转录、染色质调节、蛋白质折叠、定位和降解。
分子胶诱导泛素连接酶和POI之间的相互作用,导致POI泛素化和随后的降解。尽管分子胶和PROTAC都利用UPS降解蛋白,但它们有几个区别:1)PROTAC是异双功能降解剂、同时与E3连接酶和POI相互作用,而分子胶可以仅与E3连接酶或POI相互作用、并诱导它们之间的相互作用;2)分子胶没有连接子,比PROTAC分子量更小、口服生物利用度更高、细胞渗透性更好;3)分子胶更难设计,尽管有一些理性设计策略正在开发。
分子胶的典型例子包括沙利度胺、来那度胺和泊马度胺,早在它们的功能机制被阐明之前就已经被FDA批准。几年后,人们发现这类化合物通过充当分子胶发挥抗肿瘤活性,它们诱导E3连接酶、Cereblon(CRBN)及其转录因子底物IKZF1/3之间的相互作用,导致IKZF1/3的降解。

2.1.3 双机制降解剂
肿瘤治疗通常需要不止一个靶点,目前已经出现一种新型降解剂,拥有PROTAC和分子胶的双重特性,通过调节连接子长度来平衡PROTAC和分子胶的活性。

2.2溶酶体途径的靶向蛋白降解
溶酶体是胞内降解的主要细胞器,通过内吞、吞噬或自噬等作用接收被降解物质:内吞作用是将一些细胞表面蛋白回收到细胞膜或其他细胞器,而另一些被K63泛素链标记、并被分拣到转运所需的内体分拣复合物(ESCRT)来进行降解;吞噬作用是一种特殊形式的内吞,是细胞用于吞噬微生物病原体或其他大颗粒物;自噬作用是一种进化上保守的过程,细胞通过溶酶体清除不必要或有功能障碍的细胞器和蛋白质,将它们包裹在双层膜的囊泡中、也成为自噬体,然后与溶酶体结合并进行分解。

随着对内体-溶酶体和自噬体-溶酶体降解通路的深入研究,近年来出现了通过溶酶体途径的TPD策略,如LYTAC、AbTAC、ATTEC、AUTAC、AUTOTAC、双特异性适配体嵌合物等。与只能降解某些胞内蛋白质的蛋白酶体途径TPD相比,基于溶酶体的TPD能够清除蛋白质聚集物、受损细胞器、膜和胞外蛋白质等。

2.2.1 LYTAC
LYTAC是一种通过内体-溶酶体通路诱导细胞外和膜蛋白降解的技术,它可以同时结合膜蛋白的胞外结构域或胞外蛋白,以及位于细胞表面的溶酶体靶向受体(TLR,形成三元复合物后通过网格蛋白介导的内吞作用使得蛋白质内化,POI随后被降解。
由于这些蛋白占编码蛋白的40%,并且是神经退行性疾病、自免疾病和肿瘤的关键因素,LYTAC是PROTAC的良好补充。

2.2.2 双特异性适配体嵌合物
与LYTAC类似,双特异性适配体嵌合物也通过内体-溶酶体途径介导POI的降解;与LYTAC相反,双特异性适配体嵌合物利用靶向CI-MPR和跨膜POI的DNA适配体,将膜蛋白运送到溶酶体进行降解,同时对非靶向蛋白的水平没有显著影响。
总的来说,这种方法提供了一个强大、有效和通用的平台来诱导膜蛋白的降解,核酸适配体比抗体的制备更简单、合成更精确、也更稳定。
2.2.3 AbTAC
基于抗体的PROTAC (AbTAC)可以诱导细胞外和膜蛋白的降解。与传统的PROTAC相比,AbTAC可以靶向膜蛋白,从而大大扩展了当前TPD策略的潜在底物。虽然名为PROTAC,但AbTAC与LYTAC的关系更密切,它利用双特异性抗体,一个臂靶向细胞表面POI、另一个臂靶向跨膜E3连接酶,也通过内体-溶酶体通路来诱导POI复合物的内化和降解。
然而,AbTAC的作用机制不如LYTAC清楚,特别是不知道POI的胞内段是否在内吞作用之前被泛素化,如果是的话泛素化是如何促进复杂的内化的。

2.2.4 GlueTAC
GlueTAC利用三种主要技术来降解细胞表面蛋白:1)用纳米抗体取代传统抗体,以促进细胞渗透;2)在纳米抗体和抗原之间引入共价相互作用,以提高亲和力,并使脱靶效应最小化;3)穿膜肽和溶酶体分拣序列(CPP-LSS)与纳米抗体结合,以促进内化和溶酶体降解。
GlueTAC需要考虑几个问题:1)GlueTAC在纳米抗体中引入了非天然氨基酸,并在纳米抗体和抗原之间形成共价键,因此需要仔细评估安全性;2)纳米抗体没有重链,不能与FcRn结合;3)半衰期也需要确定。

2.2.5 AUTAC
除了内体-溶酶体通路,自噬-溶酶体通路也是TPD的另一种途径。8-硝基-cGMP是细胞中介导自噬体招募的重要信号分子,这一特性被用于开发自噬靶向嵌合体(AUTAC),它由三部分组成:基于cGMP的降解标签、连接子和POI或细胞器的小分子配体。AUTAC分子触发K63标记的多重泛素化,以及随后的溶酶体介导的降解。
除了细胞质蛋白,线粒体等细胞器也可以通过AUTAC降解,而线粒体功能障碍与许多衰老相关的疾病有关,去除功能障碍或受损的线粒体可能会改善这些疾病。

2.2.6 ATTEC
自噬体束缚化合物(ATTEC)通过将POI束缚于自噬体而发挥作用。与AUTAC招募自噬体进行降解不同,ATTEC结合自噬体的关键蛋白之一LC3。

2.2.7 AUTOTAC
自噬受体p62/SQSTM1的功能是桥接多泛素化POI和自噬体,将前者结合到p62的UBA域,导致p62的构象变化,暴露其LIR序列,并促进了它与自噬体膜上LC3的相互作用。
AUTOTAC不仅可以介导单体蛋白的靶向降解,还可以介导易聚集蛋白的靶向降解,相比之下PROTAC和分子胶则通常在处理错误折叠的蛋白质时无效。
2.2.8基于CMA的降解剂
分子伴侣介导的自噬(CMA)是三种自噬途径之一,蛋白质被分子伴侣选择后靶向溶酶体,并直接转运穿过溶酶体膜进行降解。基于CMA的降解剂包括3个功能域:穿膜序列、POI结合序列和靶向CMA序列。进入细胞后,它通过POI结合序列与靶蛋白结合,再转运到溶酶体进行降解。
基于CMA的降解剂至少有两个主要障碍,一是稳定性,二是递送效率,目前还有比较大的挑战。

3. 靶向蛋白降解的临床转化
3.1 肿瘤
已经有多个TPD分子(大多基于PROTAC)已在针对肿瘤的临床试验和临床前研究中显示出潜在的治疗效果。
PROTAC从2019年进入临床,目前已有近20个(下表截至2022年3月)。最突出的是Arvinas(ARVN)公司的两个正在临床二期的分子,分别是靶向ER的ARV-471和靶向AR的ARV-110;其他主要玩家包括BMS、Nurix、Kymera、C4等公司,主要靶点包括STAT3、BTK、BCL-XL、EGFR-L858R等。中国公司在这种又火又看似门槛不高的风口上自然也不会旁观,百济神州(BGNE)海思科(SZ002653)海创开拓等一票人马也都进入临床了。
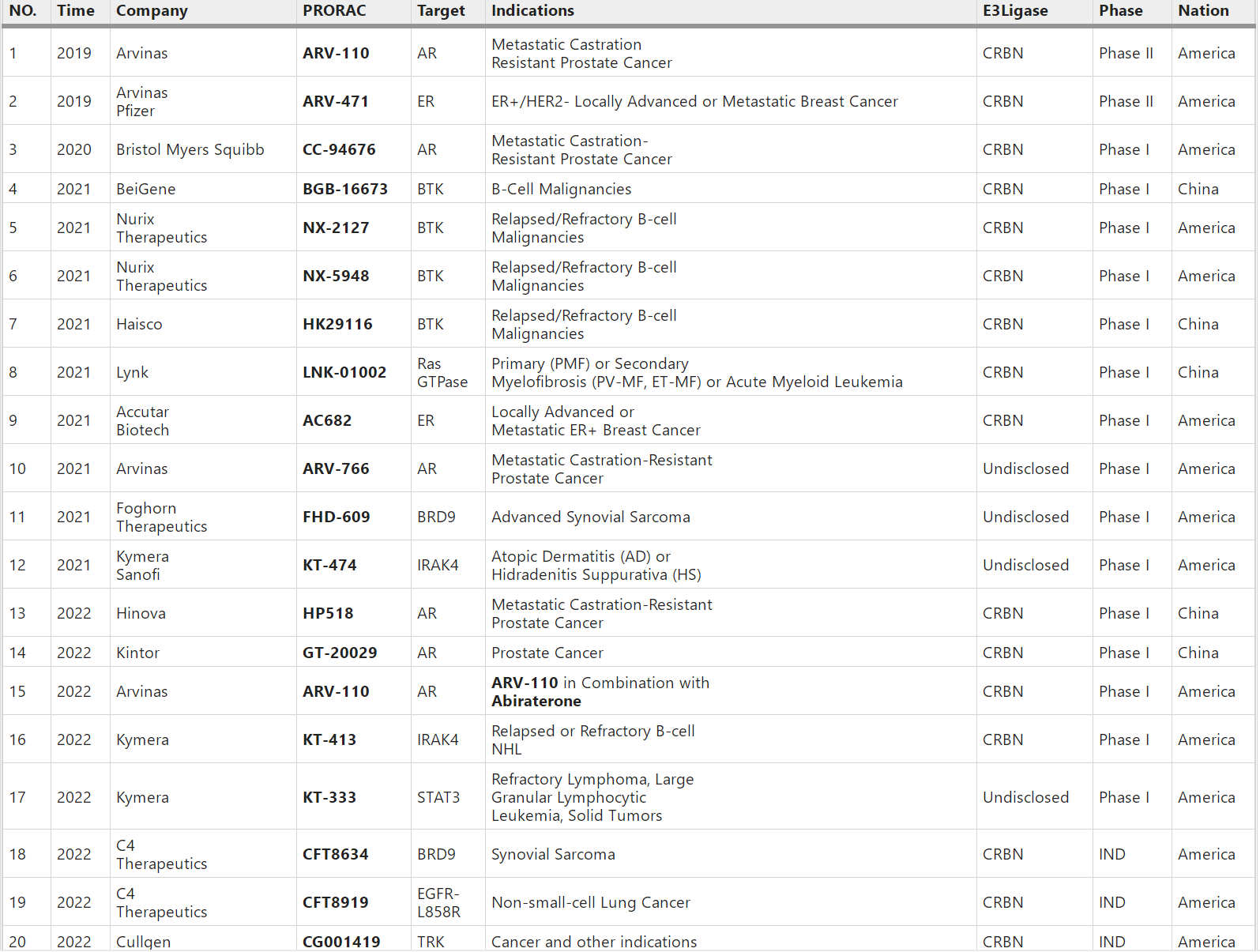
分子胶也已进入临床阶段,基本都是基于新一代CRBN,靶向IKZF1/3和GSPT1等。最突出的是BMS的CC-90009和CC-92480,还有C4和Novartis等公司。

3.1.1 ARV-471
这是一款靶向雌激素受体的PROTAC降解剂,单药正在临床试验中用于治疗ER+/HER2-的局部晚期或转移性乳腺癌。
ARV-471的临床1期数据显示了强有力的有效性,对末线患者有40%的临床受益率,且优于氟维司群和其他临床阶段的SERD。目前已进入单药临床2期,以及CDK4/6抑制剂联用的临床Ib期。


3.1.2 ARV-110
这是一款靶向雄激素受体的PROTAC降解剂,单药正在临床试验中用于治疗末线mCRPC患者。
ARV-110的临床1期数据显示,该药的耐受性良好、成功介导了肿瘤中靶蛋白PSA的降解、也显示出抗肿瘤活性,这也是PROTAC分子在首次在人体中展示出POC证据。目前已在准备临床3期。


3.2 神经退行性疾病
神经退行性疾病是一组以进行性运动或认知障碍为特征的疾病,包括阿尔茨海默病、帕金森病和亨廷顿病等,与蛋白质错误折叠形成的不溶性聚集体密切相关,这些错误折叠的蛋白质通常表现出与其正常功能无关的异常蛋白质相互作用。
目前开发出一种靶向tau蛋白的PROTAC,采取嵌合体结构,由tau结合肽、连接子、VHL结合肽和穿膜肽组成,能导致tau蛋白的显著降解和Aβ神经毒性的降低。
3.3 炎症性疾病
目前已有多种靶向IRAK-4的PROTAC分子,其中一种已进入治疗自免疾病的一期临床。IRAK-4是IRAK激酶家族的成员,参与TLR和IL-1R信号通路调控,TLR激活后IRAK-4被招募形成Myddosome复合物,随后导致IRAK1/2等的磷酸化。与常规抑制剂相比,IRAK-4降解剂通过消除IRAK-4的功能提供了巨大的优势。
另有两种靶向BTK的PROTAC正在肿瘤和自免疾病的一期临床中。BTK是炎症和肿瘤的已知靶点,虽然已有BTK抑制剂获批,但BTK突变限制了有效性,这可以由BTK降解剂来解决,因为它们可以降解野生型和突变型BTK蛋白。
3.4 抗病毒
TPD可能代表一种新的抗病毒治疗方法。目前已在尝试用于丙型肝炎病毒(HCV)NS3/4A蛋白酶的降解,基于蛋白酶抑制剂Telaprevir的PROTAC可以在细胞模型中抑制HCV。除了PROTAC,自噬-溶酶体途径的AUTAC和ATTEC,也可以用于消除关键的病毒蛋白。
4. PROTAC的建构设计
回顾PROTAC技术发展过程中,正确选择降解目标蛋白是关键,而选择肿瘤细胞特异性的E3连接酶能够保障其治疗窗口。
4.1 靶点选择
最初一波进入临床的TPD分子都针对的是传统的药物靶点,它们已经经过临床验证且较容易合成。通过这些low-hanging fruit,对PROTAC的机制形成了较好的验证,然而这种疗法的真正前景是解决目前用现有疗法难以成药的靶点。
PROTAC治疗的最佳靶点筛选原则,有几个共同特征,包括:1)通过过表达、突变、聚集、异构或定位等偏离自然状态,会导致疾病引起;2)有E3连接酶可以接近的结合面;3)有用来穿入蛋白酶体的非结构化域;4)对靶向治疗产生抗性突变;5)具有支架功能;6)用其他方式“不可成药”。

在目前处于临床前开发阶段的PROTAC靶点中,最常见的是AR,其次是BRD4、EGFR和ALK等,大多数都是针对转录调节因子和具有激酶活性的蛋白而设计。

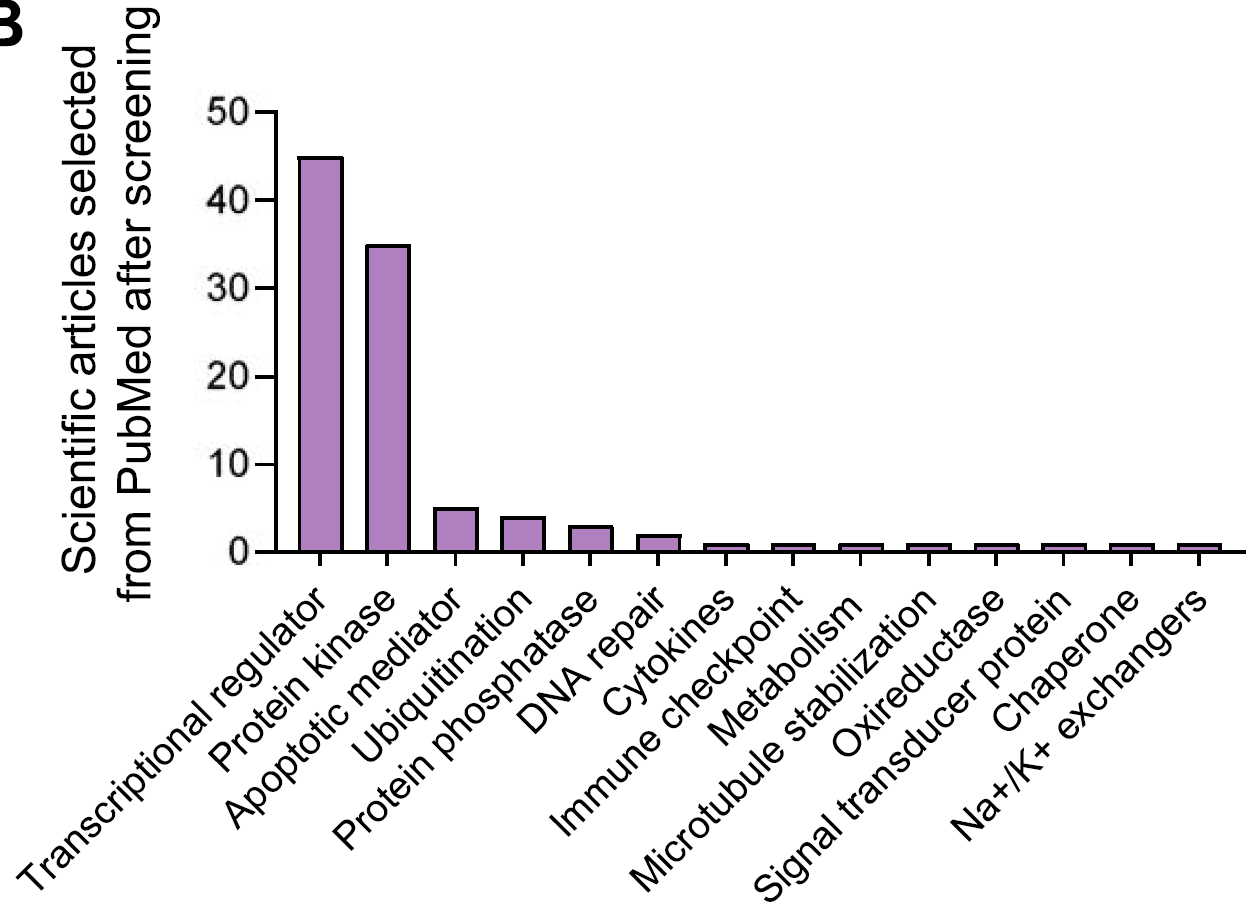

通过分析CRISPR和RNAi敲除基因对细胞存活的影响,得出选择性较高的靶点名单(下图A),并在五种最常见的肿瘤中进行CERES和DEMETER2评分(下图B),发现如CDK9、AURKA、PLK1以及BCL2、MCL1、PTPN11、BRD4、PTK2等基因,在CRISPR和RNAi方法之间结果有差异。

进一步研究这些靶点高表达肿瘤的RNA水平,发现BCL2、CDK4和MCL1在所有上述肿瘤中都表达较高,其中MCL1尤其在乳腺癌和肺腺癌中高表达,CDK4尤其在结肠腺癌中高表达。

在不同器官的正常组织中,MCL1普遍表达较高、尤其是肺中,其他靶点诸如BCL2L1、CDK4和CDK9也表达较高。

4.2 连接酶选择
在目前处于临床前开发阶段的PROTAC连接酶中,最常见的是CRBN,其次是VHL和IAP。
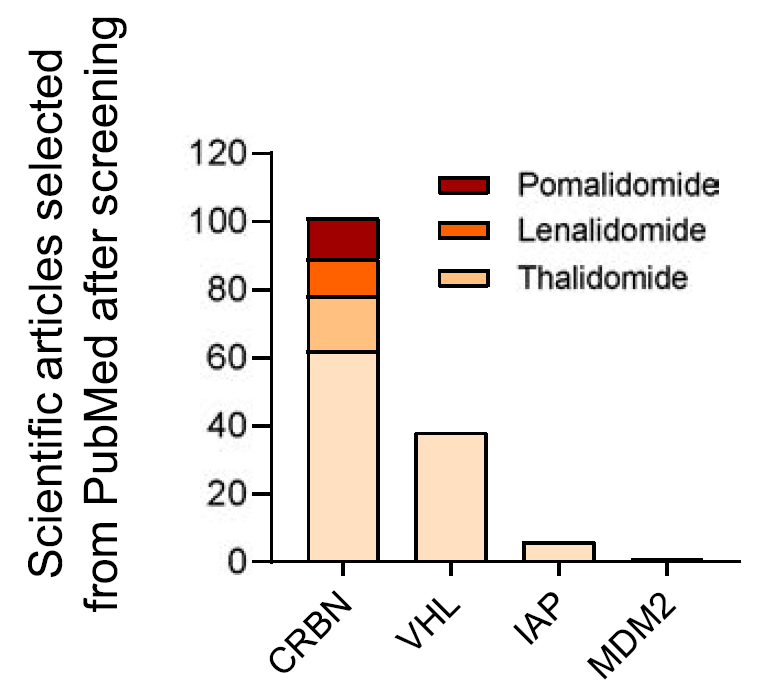
通过分析肿瘤中RNA水平,发现大多数E3连接酶在肿瘤中都表达并不高,只有MDM2在乳腺癌、肺癌、前列腺和胃癌中较高。

在不同器官的正常组织中,MDM2普遍表达相对较高,其次是cIAP和CRBN,而VHL和XIAP普遍表达偏低,但值得注意的是VHL在骨髓中表达尤其高。
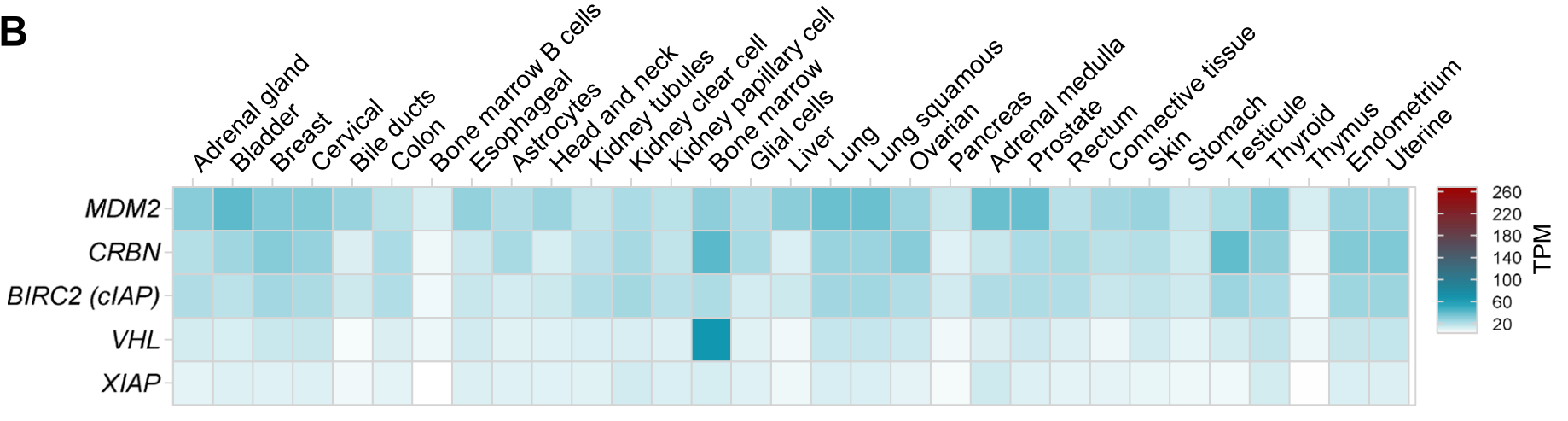
尽管CRBN和VHL已经很常用,但开发新E3连接酶的时机也已成熟。新连接酶需要具有组织和细胞类型的特异性、肿瘤靶向性等。目前已鉴定出一些新的连接酶,包括在骨骼肌中的KLHL40/41、中枢神经系统中的RNF182和TRIM9、以及FBXO44等。

4.3 靶点和连接酶综合考虑
靶点和连接酶都高表达的肿瘤,主要包括MDM2连接酶与BCL2、BRD4、CDK9、CDK4、PLK1、MCL1和PIK3C3等的组合在胃腺癌和乳腺癌中。

在正常组织,所有器官整体MDM2与CDK4、CDK9、BCL2、MCL1的组合表达都较高,而骨髓中涉及到的表达最高、特别是CRBN和VHL连接酶。
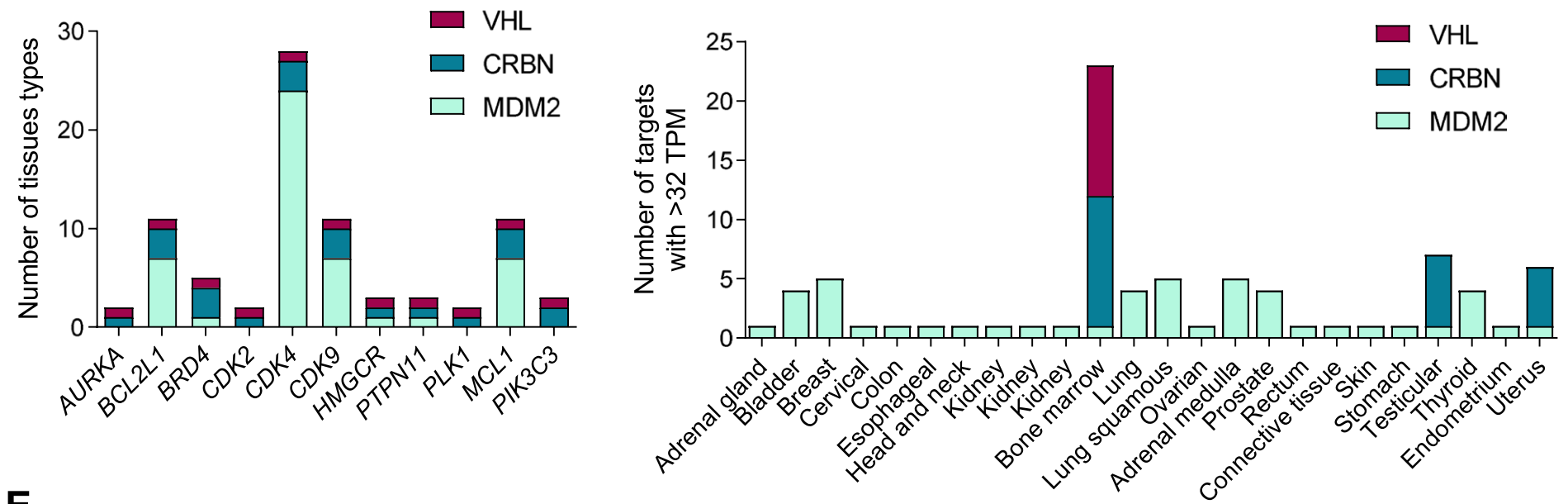
最后将靶点和连接酶的组合,考虑在肿瘤中高表达且在正常组织中低表达,MDM2连接酶与胃癌中的BCL2L1、BRD4、CDK9、PLK1、MCL1等靶点,以及MDM2连接酶与乳腺癌中的PIK3C3靶点,都是比较有吸引力的选择。

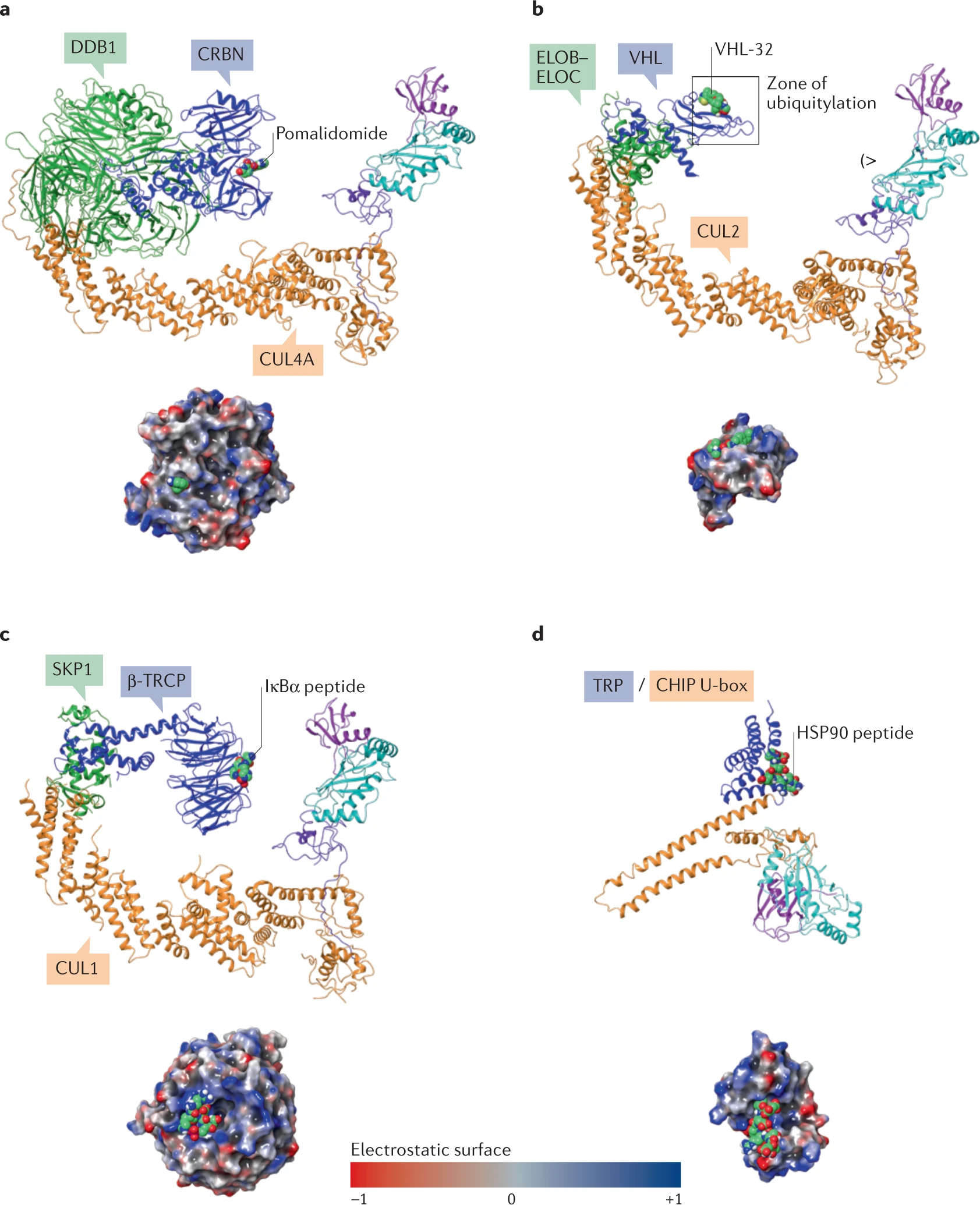
4.4连接子
连接子可用于调节PROTAC的分子量和属性,而绝不仅是将POI和E3连在一起并影响性能,它可以与PROTAC的构象和降解效率相关,并影响E3-POI相互作用和泛素化区内POI的递呈。
5. 未来展望
自2001年报道第一个PROTAC分子以来,TPD技术正在从概念进入实际应用阶段。虽然这个领域取得快速发展,但仍有许多挑战有待解决,主要来自两个方面
5.1 分子设计和成药性
靶蛋白目前多数PROTAC都采取靶向激酶的策略,很少有靶向不可成药靶点的例子。靶向激酶的PROTAC分子通常通过使用现有的小分子抑制剂作为靶蛋白配体进行修饰来获得,而这些抑制剂是为靶向激酶结合口袋设计的。对于不可成药的靶点,包括转录因子、磷酸酶、蛋白质之间相互作用等,由于缺乏有效的小分子配体,总体进展缓慢。
目前仍然很少出现可用的新E3连接酶,如何扩展可用于PROTAC技术的E3泛素连接酶也是面临的明确挑战之一。
连接子可以深刻影响PROTAC分子的活性、选择性以及成药性,而如何有效设计连接POI和E3连接酶的连接子,也是重要的问题。
由于分子量大,PROTAC分子作为药物的药理性质通常不太好,如何快速进行分子优化是一个巨大的挑战。
PROTAC分子通常具有“钩子”效应,能否用合理的分子设计来削弱甚至消除这种浓度依赖问题?显然,传统的药物化学筛选和优化方法不适合PRTOAC分子的快速开发和优化,尤其是对于不可成药的靶点。
针对上述问题,AI技术(蛋白结构预测)、虚拟药物筛选技术和DEL筛选技术等,可以帮助开发靶点和E3连接酶的相应配体。基于这些筛选的配体,有必要发展高效的合成方法,快速有效地构建以骨架多样性为特征的大规模PROTAC分子库,用于高通量筛选和优化分子成药性。在分子设计方面,现有的PROTAC分子与POI和E3连接酶蛋白的三元复合物结构还很少,未来通过X射线或冷冻电子显微镜获得的更复杂的信息将有助于更好的分子设计。近年来,alphafold2预测蛋白质和相关复杂结构的能力的突破也可能有助于PROTAC的设计。对于分子优化,增加分子的整体刚性和降低分子量,一般可以改善HIT化合物的性质,如口服生物利用度和ADME等。
5.2 生物活性评价
现有的分子筛选技术,主要依靠免疫印迹法和蛋白质组学方法,不仅费时、费力、效率低,而且成本高。近年来,逐渐引入荧光标记和HiBiT等筛选技术逐渐被引入,未来还需要更多新的高通量和高灵敏度的方法来进行快速和准确的评估。
除了常规的评价指标如溶解度、体内外活性、毒性和其他药学性质,由于PROTAC的作用是催化循环反应,因此传统方法无法准确评估PROTAC的PK和PD性质。对于这种新的药物形式,非常需要开发与蛋白质降解更一致的PK/PD模型。
如何更好地理解PROTAC分子的降解活性和可降解性、选择性、脱靶效应、药理作用(基于不同靶点/细胞系/动物模型),以及在临床治疗中如何相应地实现区分,都是较大的挑战。
以上这些问题目前都没有现成的答案,但我们相信,随着更多的生物学、药理学和临床研究的发展,新的评价方法和系统将逐步建立起来以解决这些问题。相信在未来,越来越多的靶向蛋白降解药物分子不仅可以作为基础生物学研究的工具,还将进入临床,解决患者的实际需求。
附:参考文献

网页链接

网页链接

网页链接

网页链接
欣荷:
PROTAC
海创药业-U(SH688302)
海思科(SZ002653)
股票
MORE>
- 最近发表
- 标签列表
-
-
SZ002278 SZ002624 SH603367 SZ300100 SZ002963 SH605589 SH600378 SH603208 SZ300505 SZ002558 SH603069 SH605305 SZ000711 SZ002786 SH601618 SH688575 SH600027 SZ300151 SH600869 SZ000005 SZ300783 SZ300124 SH600058 SH600390 SH600757 SH688271 SZ002668 SH603650 SZ003031 SH601818 SZ002614 SH603040 SZ300427 SH600884 SH603555 SH600721 SH688066 SZ000700 SH605167 SZ300879 SH603158 SH603486 SH603678 SZ002494 SZ002504 SZ002714 SH600011 SZ002266 SZ300292 SZ000821